






















 精确搜索
精确搜索 我的草稿
我的草稿
 我的购物车
我的购物车
 精确搜索
精确搜索

技术 | qPCR不正常曲线分析
溶解曲线主峰左侧有峰

一般考虑是引物二聚体或者短的非特异性扩增,解决手段通常有:
◆ 降低引物浓度
20ul体系正常参考引物用量是10uM浓度 0.4ul
◆ 提高退火温度
梯度摸索引物退火温度,一般退火温度不建议超过63°C
◆ 增加模板浓度
当目的基因表达量极低时,染料法QPCR容易形成引物二聚体产物影响实验结果,提高模板浓度
◆ 重新设计引物
溶解曲线主峰右侧有峰
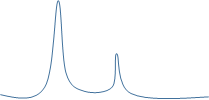
一般考虑非特异性扩增,去除非特异性扩增的手段有:
◆ 提高退火温度:
梯度摸索引物退火温度,一般退火温度不建议超过63°C
◆ 提取的RNA需要去除基因组DNA污染,或者
◆ 重新设计引物
ROX添加不当
ROX矫正不当可能形成如下熔解曲线图
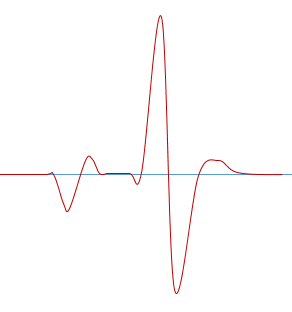
解决手段通常有:
◆ 一定要清楚机器的ROX矫正信息,加的时候一定要看清标签,高低要分清
◆ ROX要完全融化,混合均匀后再使用
◆ 如果是需要矫正的机器型号但是用了不校正的染料,那么可以关闭软件里面的ROX矫正选项,重新分析数据
高浓度抑制
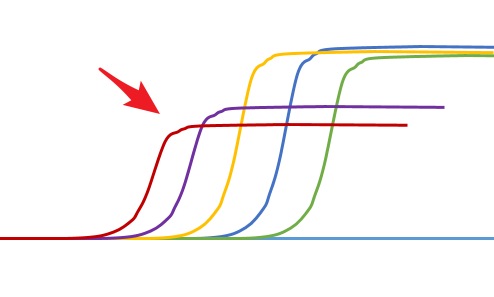
◆ 高浓度模板可能存在一些抑制因素,导致扩增产物的减少,稀释后抑制就降低或者解除了
◆ 所以如果出现这样的情况可以调整模板浓度,Ct滞后我们也可以调整下稀释倍数,保证足够多的标准点。
浓度梯度Ct值异常

◆ 前面不能出现梯度,原因可能是模板量过高,超出了线性范围,那么需要去掉前面不能分开的梯度,选择后面梯度;
◆ 后面不能出现梯度,原因可能是模板量低,超出了线性范围,那么需要去掉后面这些梯度,重新摸索高浓度梯度。
无扩增曲线
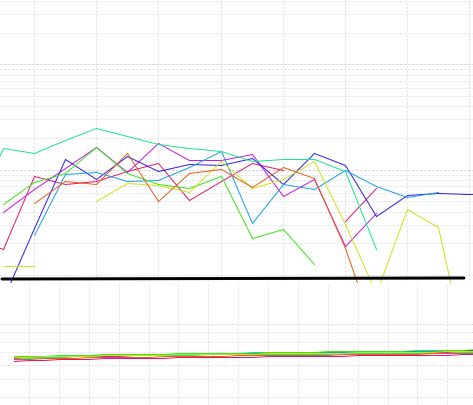
◆ 机器设定:首先要看下程序的设定方面,如果程序设定错误,那么是采集不到荧光信号的,
(两步法扩增程序一般将信号采集设置在退火延伸阶段;三步法扩增程序应当将信号采集设置在72°延伸阶段)
◆ 循环数:循环数不够,一般要设置40个循环,但是循环数太多的话会增加背景信号,降低数据的可信度;
◆ 引物降解:可以通过变性PAGE检测引物是否降解
◆ 引物不合适:重新设计引物
◆ 模板浓度太低: 减少模板稀释倍数,如果是未知浓度样品建议从高浓度开始预实验;
◆ 模板降解:重新制备模板,重复实验
◆ 表达量低:重新反复设计引物,4-5对不可以放弃该基因
Ct值偏大
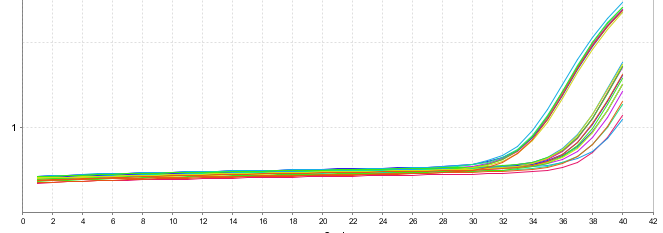
◆ 引物不够好,扩增效率低,重新设计引物或探针;
◆ 优化PCR条件:
- 改用三步法;
- 增加镁离子浓度(如果用mix应该也调整不了这个浓度)
- PCR各反应成分的降解或加样量不足
- 扩增片段太长:建议长度80-200bp
◆ 基因表达量低:
若重新反复设计引物仍不能解决问题,可以放弃该基因或使用其他更为灵敏的检测方法
扩增曲线先下降后上升

◆ 模板浓度太高,基线的终点值大于CT值:
手动设置减小基线终点值,重新分析数据,(默认的基线范围一般是3~15个循环,如果遇到扩增在10个循环就起来的那么收到把基线范围设置到3~5就可以了,或者是CT值前4~5个循环,基线的设置很可能会影响到引物的扩增效率的结果)
没有对数增长期
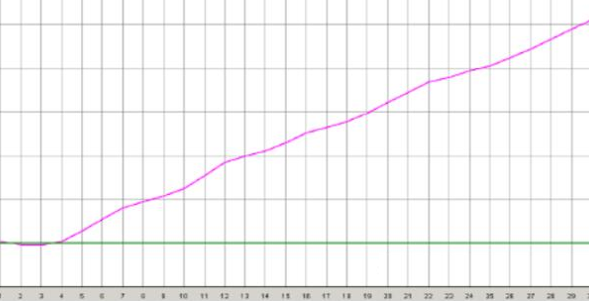
◆ 探针部分降解导致的,如反复冻融探针;在光下暴露时间过长都可能引起,需要重新更换引物探针
◆ PCR反应液中有PCR抑制物,如模板浓度过高,模板中有杂质等
某一空的荧光信号 特别强
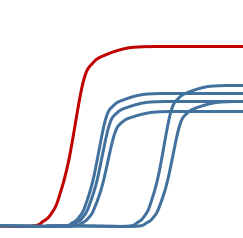
◆ 试剂制备时没有充分混匀,各管成分不一样
◆ 如果是探针法的话,可能是探针没有融化,有一管中的探针量太多
◆ 也可能是机器原因,不过这个可能性不大
扩增曲线有向上的尖峰
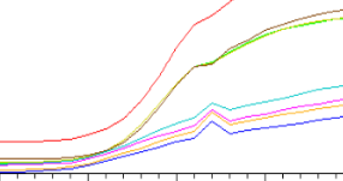
◆ 可能是电压不稳定导致的,也可能是断电或者突然开盖导致的。
山峰形扩增曲线

◆ 反应液蒸发,可能管子没有盖好或者管子质量差
标曲标准偏差大于0.5
扩增反应的对数线性期的斜率可用于检测反应效率。
● 良好的反应效率应在90%-110%之间。
● 扩增效率低于100%,是因为PCR扩增体系中的抑制因素 ,或者RNA有所降解;
● 高于100%是因为非特异性扩增或引物二聚体造成。
解决方案:
◆ 试剂使用前需要彻底融化充分混匀,包括Mix、引物和探针
◆ 使用精确的移液器,加样时候要垂直加样

● 扩增信号太弱
● 仪器本身的问题,可能在扩增过程中仪器出现了波动